Researchers have captured the first atomic-scale evidence that copper oxide catalysts can repeatedly switch between chemically active and inactive states while operating under constant reaction conditions. The work, led by Prof. Xianhu Sun and colleagues and published in the Proceedings of the National Academy of Sciences (PNAS) under the title “Oscillatory redox behavior in oxides: Cyclic surface reconstruction and reactivity modulation via the Mars–van Krevelen mechanism”, was conducted primarily at the State University of New York at Binghamton and University of Chinese Academy of Sciences, with contributions from several international research institutions.
The study reveals that copper oxide surfaces undergo rhythmic cycles of oxygen loss and replenishment, producing a self-sustaining oscillation in surface chemistry. These changes were visualized directly using in situ environmental transmission electron microscopy, allowing scientists to watch atoms rearrange in real time during hydrogen reactions. The findings provide new insight into catalyst surface reconstruction, redox cycling, and the Mars–van Krevelen mechanism, which underpins many industrial oxidation processes.
Why catalyst surfaces rarely stay still
Catalysts are often imagined as static materials that speed up chemical reactions. In reality, their surfaces constantly adapt to surrounding conditions. When gases such as hydrogen interact with metal oxides, surface atoms can move, bonds can break, and oxygen atoms can be removed or replenished. These microscopic changes can dramatically alter catalytic activity.
The PNAS study shows that copper oxide is far from chemically passive. Under hydrogen exposure at elevated temperature, the surface repeatedly toggles between two structural states. One is oxygen-rich and highly reactive. The other is oxygen-deficient and temporarily inactive. This switching behaviour does not require changing temperature or gas composition. Instead, it emerges naturally from the interplay between surface chemistry and oxygen transport inside the material.
This behaviour is particularly important for reactions governed by the Mars–van Krevelen mechanism, in which lattice oxygen from the catalyst directly participates in chemical transformations. Such reactions are central to hydrogen processing, methanol synthesis, and environmental catalysis. Understanding how surfaces evolve during these reactions has remained a major challenge until now.
Watching atoms move in real time
To capture these processes, the research team used environmental transmission electron microscopy. This technique allows materials to be imaged at atomic resolution while exposed to reactive gases and high temperatures. In the experiment, copper metal was first oxidized to form copper oxide. The environment was then switched to hydrogen gas while maintaining a temperature of around 300 degrees Celsius.
As hydrogen reacted with the oxide surface, the microscope recorded dynamic changes at the atomic level. The researchers observed that the surface flattened and reorganized into two distinct configurations with different oxygen contents. These configurations repeatedly converted into one another in a cyclic pattern.
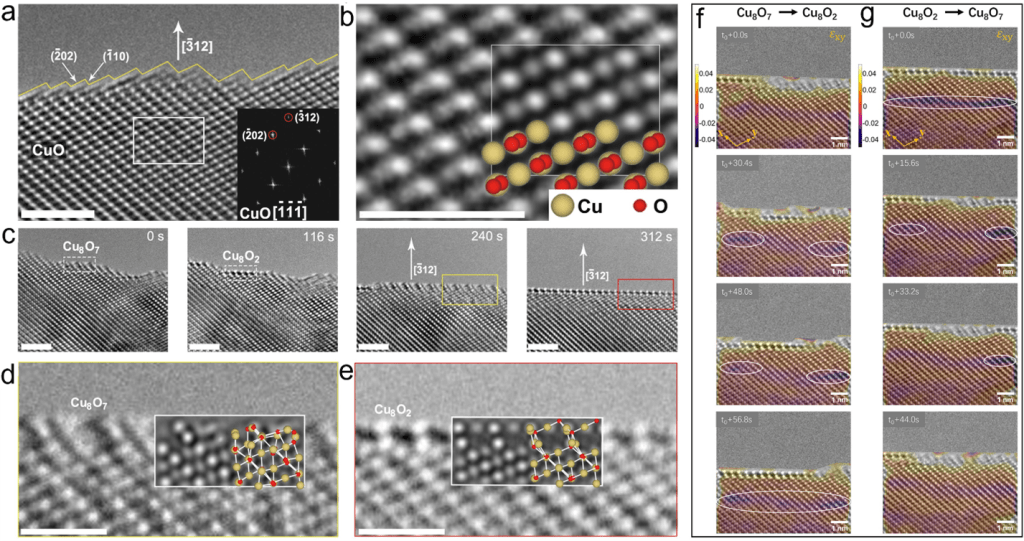
What makes this observation remarkable is the direct visualization of surface reconstruction during a catalytic reaction in Fig. 1. Previous studies inferred such behaviour indirectly from chemical measurements. Here, atomic-scale restructuring could be observed frame by frame, providing unprecedented evidence of oscillatory redox behaviour in an oxide catalyst.
Two faces of copper oxide
The study identified two dominant surface states on copper oxide. One configuration was relatively oxygen-rich and dominated by copper ions in a higher oxidation state. This surface readily activated hydrogen molecules and promoted the formation of water by reacting hydrogen with lattice oxygen. As water molecules desorbed from the surface, oxygen vacancies formed in the crystal lattice.
The second configuration was oxygen-deficient and characterized by copper ions in a lower oxidation state. This surface showed much lower chemical reactivity. Once the surface reached this state, hydrogen adsorption slowed dramatically, effectively placing the catalyst into a temporary inactive phase.
The role of the Mars–van Krevelen mechanism
At the heart of this behaviour lies the Mars–van Krevelen mechanism. In this well-known catalytic pathway that proposed about 70 years ago, oxygen atoms from the solid catalyst participate directly in chemical reactions. After being removed from the surface, these oxygen atoms must be replenished from the bulk of the material for catalysis to continue.
The new research demonstrates that this replenishment process is slower than oxygen removal during hydrogen reactions. This time lag creates a separation between the reduction and reoxidation steps. As a result, the surface alternates between states that favour oxygen extraction and states that promote oxygen recovery.
This spatiotemporal separation of redox processes creates a feedback loop. When the surface becomes oxygen-rich, it is chemically active and rapidly loses oxygen. When it becomes oxygen-deficient, it becomes chemically inert, allowing oxygen to diffuse from the subsurface to restore activity. The cycle then repeats without any external intervention.
Oxygen diffusion is the rate-limiting factor
To understand why this oscillation occurs, the researchers combined microscopy with density functional theory calculations. These simulations revealed that the slowest step in the process is the upward diffusion of oxygen atoms from the subsurface region to the surface. While oxygen can move relatively easily within the bulk of the material, crossing the final atomic layers to reach the surface requires overcoming a significant energy barrier.
This bottleneck creates a delay between surface oxygen loss and replenishment. During this delay, the catalyst remains in its oxygen-deficient, low-activity state. Once enough oxygen reaches the surface, the oxygen-rich configuration reforms, and the surface becomes reactive again.
The modelling results aligned closely with experimental observations. Regions of compressive strain were detected beneath the surface during oxygen depletion, indicating the accumulation of oxygen vacancies. These strain patterns relaxed once oxygen returned to the surface, further confirming the cyclic nature of the process.
Why copper oxidation states matter
Another important finding concerns the oxidation state of copper ions at the surface. Cu2+ ions, similar to bulk copper oxide, dominated the oxygen-rich surface configuration. This state promotes strong hydrogen bonding and facilitates the formation of water.
In contrast, the oxygen-deficient surface was dominated by Cu1+ ions. This change in the electronic structure reduced the surface’s ability to activate hydrogen molecules. As a result, hydrogen adsorption became energetically unfavourable, reinforcing the inactive phase of the cycle.
These oxidation state transitions provide a clear link between surface chemistry and catalytic performance. They show how subtle electronic changes at the atomic level can determine whether a catalyst is highly active or temporarily dormant.
A broader message for materials science
Although this study focused on copper oxide, the authors emphasize that the underlying mechanism is likely to apply to many other oxide materials. Any catalyst that relies on lattice oxygen participation may exhibit similar surface reconstruction and redox oscillations. Techniques that combine real-time imaging with theoretical modelling are becoming essential tools for revealing how functional materials behave at the atomic level.
Reference
Sun, X., Wu, D., Wang, J., Patel, S. B., Zhu, W., Yang, J., Yang, T. T., Ye, S., Chen, X., Zhu, Y., Qiao, L., Li, M., House, S. D., Su, J., Said, W. A., Boscoboinik, J. A., Yang, J. C., Sharma, R., & Zhou, G. (2025). Oscillatory redox behavior in oxides: Cyclic surface reconstruction and reactivity modulation via the Mars–van Krevelen mechanism. Proceedings of the National Academy of Sciences, 122(24), e2422711122. https://doi.org/10.1073/pnas.2422711122