
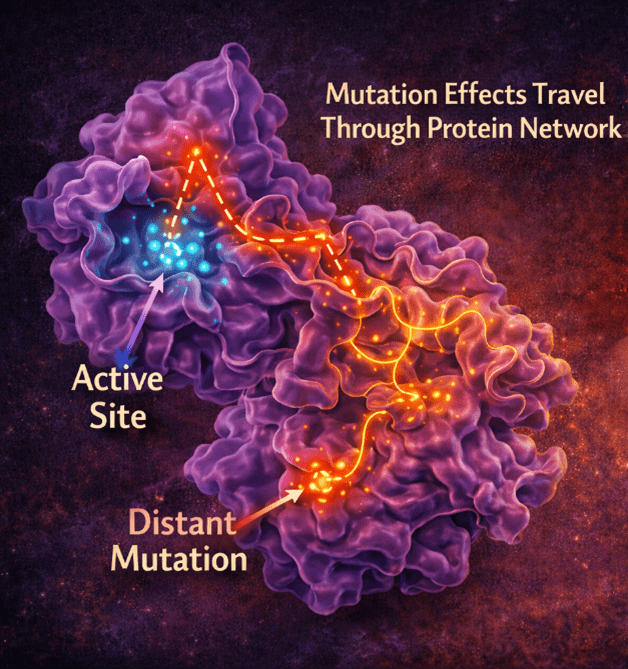
A new computational study published in Physical Chemistry Chemical Physics Journal by researchers at the Indian Institute of Technology Kharagpur reveals how subtle genetic mutations in a thyroid enzyme can disrupt its function in unexpected ways. The findings highlight how one mutation can cause major structural destabilisation, while another, located far from the active site, quietly reshapes the enzyme’s internal environment through long-range interactions. These changes weaken key molecular alignments required for efficient electron transfer, ultimately impairing thyroid hormone production.
For researchers and clinicians studying congenital hypothyroidism, this work may demonstrate that a detailed molecular picture can guide understanding, revealing that disease mechanisms arise from the full dynamic behaviour of proteins, not just their active sites.
A tiny molecular glitch with big consequences
What if a mutation far away from where the action happens could still break a crucial biological process? This is exactly what the authors found in a key human enzyme involved in thyroid hormone production. Even subtle genetic changes, some sitting far from the enzyme’s active site, can disrupt how it works, leading to congenital hypothyroidism, a condition affecting about 1 in 3000 newborns.
The study reveals that the molecular-level story is more complex than scientists once believed; it’s not just where a mutation occurs, but how its effects ripple across the entire protein.
The iodine recycler, your body depends on
Thyroid hormones rely on iodine, a scarce but essential element. To conserve it, the body uses an enzyme called iodotyrosine deiodinase (hIYD), a molecular recycler that salvages iodine from partially used building blocks. When this enzyme fails, iodine is lost instead of being reused. The result can be:
- Reduced thyroid hormone production
- Goitre (thyroid enlargement)
- Developmental and cognitive delays if untreated
Several mutations in the gene encoding this enzyme (DEHAL1) have been identified as disease-causing. But until now, the molecular-level behaviour of these mutations has remained largely unknown.
How the study was conducted
The researchers used a combination of advanced computer simulations to understand how small genetic mutations affect the behaviour of the hIYD protein.
First, the researchers improved the modeling of iodine-containing molecules by adding a special “extra-point” charge. This helps capture the unique way iodine atoms interact with their surroundings, leading to more realistic simulations.
Next, they analysed how different parts of the protein communicate with each other. By turning the protein into a network, where amino acids act like connected nodes. They could track how information flows across the structure. This revealed which regions are most important and how mutations disrupt these internal communication pathways.
To understand how strongly molecules bind to the protein, the team calculated binding energies using a method that combines physical interaction forces with solvent effects. They also broke these energies down to individual amino acids, helping identify key “hotspots” responsible for binding.
Finally, they used a statistical model to study protein stability. This approach treats each part of the protein as either folded or unfolded and calculates how mutations shift the overall balance. It provides insights into how mutations can make the protein more or less stable.
Not all mutations are created equal
The researchers studied three disease-causing mutations: Arg101Trp (near the active site), Phe105–Ile106Leu (a small deletion in a critical flavin binding loop), and Ile116Thr (located far from the active site).
At first glance, one might expect mutations closest to the active site to be the most damaging. However, using long-timescale molecular simulations, it is observed that:
- The Phe105–Ile106Leu deletion causes the most severe structural disruption
- Arg101Trp weakens key molecular interactions needed for enzyme function
- Ile116Thr, despite being distant, subtly alters the protein’s internal environment
This challenges the traditional view that mutations act only locally.
Proteins behave more like networks than machines
Proteins are not rigid objects; they are dynamic, interconnected systems. A change in one region can influence distant parts through what scientists call long-range coupling. Some mutations weaken critical interactions that hold the enzyme together, while others alter how the protein “breathes” and fluctuates.
Even distant mutations can change internal hydration patterns slowly and side-chain conformations, leading to a delayed onset in patients. In particular, the deletion mutation (Phe105—Ile106Leu) disrupted the enzyme’s dimer interface, the region where two protein units come together, weakening its overall stability.
When folding goes off track
Mutations occur in genes, which encode proteins. Before a protein can function, it must fold into the correct shape. The mutations don’t just affect the final structure; they can change the pathway the protein takes to get there. The normal enzyme (wild-type) follows a coordinated folding route, while mutations can reroute this process, creating unstable intermediate states.
The point mutation (Arg101Trp) in the active site of hIYD increases the energetic difficulty of folding by introducing a higher-energy intermediate. In contrast, the other two mutations (Phe105–I106L and I116T) reroute the folding pathway, leading to a sequence of folding events that differs from the wild type. These alterations in the folding landscape remain closely linked to the structure–function relationships and reflect subtle changes in the relative substrate-cofactor orientation within the active-site environment.
Substrate-cofactor electronic communication
At the heart of this enzyme lies a chemical reaction that depends on precise alignment between two components:
- A cofactor called flavin (flavin mononucleotide)
- A substrate molecule (iodotyrosine)
For the reaction to work, these must be positioned just right.
The mutations misalign these components by small but critical amounts. They flatten the butterfly-like structure of the flavin molecule, reducing its reactivity and weakening the electronic interactions required for efficient chemistry. Even subtle geometric changes can slow down electron transfer, the core process that drives flavin-mediated reductive dehalogenation, a rare and remarkable reaction in nature.
One of the most intriguing findings involves the Ile116Thr mutation, located far from the active site. This mutation leaves the overall structure mostly intact, but increases flexibility and water exposure in the protein core. This may explain why patients with this mutation often develop symptoms later in life. The enzyme might work, just not robustly, especially under stress.
In this work, we decode mutation effects, not just observe them – linking structure, folding, and catalysis to reveal how and when disease truly emerges.
— Sabyashachi Mishra
Why does this matter beyond one disease?
Understanding how mutations affect proteins at this level has wide implications. Disease-causing mutations can act through unexpected, long-range effects. Subtle structural changes can have major biological consequences. Protein stability, folding, and dynamics are just as important as active-site chemistry.
This insight could help: (i) Improve the genetic diagnosis of thyroid disorders, (ii) Guide drug design targeting enzyme stability, (iii) Inform how we interpret mutations across many. diseases
Reference
Karmakar, S., Giri, B., & Mishra, S. (2026). Structural and catalytic consequences of active-site vs. distal mutations in human dehalogenase: insights from molecular dynamics simulations. Physical Chemistry Chemical Physics, 28(12), 7318-7330. https://doi.org/10.1039/D5CP04422G





