

For decades, scientists have relied on classical supercomputers to simulate the behaviour of molecules. These simulations are critical for drug discovery, materials science, and energy innovation. Yet even the fastest machines struggle when tasked with capturing the quantum behaviour of electrons. The reason is simple but daunting: electrons are governed by the rules of quantum mechanics, which are notoriously difficult to approximate.
Classical algorithms can approximate molecular interactions up to a point, but the complexity grows exponentially with system size. This limitation prevents accurate predictions for large molecules or strongly correlated materials. For researchers trying to design next-generation batteries, catalysts, or medical therapies, these computational barriers remain a serious obstacle.
How quantum computing could help
Quantum computers, unlike classical ones, are built on qubits. Qubits can exist in superpositions and become entangled, allowing them to represent complex molecular states with far greater efficiency than traditional bits. In principle, a quantum computer could model molecular interactions directly, without resorting to approximations that slow down or distort results.
This potential has drawn enormous interest from the fields of quantum chemistry and condensed matter physics. But the reality is more complicated. Current quantum computers, often called Noisy Intermediate-Scale Quantum (NISQ) devices, are limited by hardware noise, short coherence times, and fragile qubits. These machines cannot yet handle large-scale simulations with sufficient accuracy.
The bottleneck of molecular Hamiltonians
A major challenge lies in dealing with the Hamiltonian of a molecular system. The Hamiltonian is a mathematical operator describing the energy of a system. When translated into the language of qubits, it explodes into thousands of Pauli strings that must be measured separately. The measurement burden quickly becomes unmanageable, with costs scaling as O(N⁷/ε²) for a system of N qubits and accuracy ε.
Even small molecules such as hydrogen, lithium hydride, or water require substantial computational overhead when tackled with traditional variational quantum eigensolvers (VQEs). Moreover, noise in quantum circuits makes the measurements prone to errors, with deviations reaching 20 to 40 percent in some cases.
A breakthrough from IIT Kharagpur
In a paper titled SHARC-VQE: Simplified Hamiltonian Approach with Refinement and Correction enabled Variational Quantum Eigensolver for Molecular Simulation, researchers from the Indian Institute of Technology, Kharagpur present a method to overcome this barrier. The first author, Harshdeep Singh, working with co-authors Sonjoy Majumder and Sabyashachi Mishra, introduces SHARC-VQE, a novel quantum algorithm designed to cut costs and reduce errors in molecular simulations.
The method partitions the Hamiltonian into a manageable “partial Hamiltonian” containing the easy-to-execute terms, while the more difficult terms are approximated by refined operators. These refinements are then corrected at the end of the calculation. By avoiding the need to measure every Pauli string directly, SHARC-VQE makes simulations both faster and more accurate.
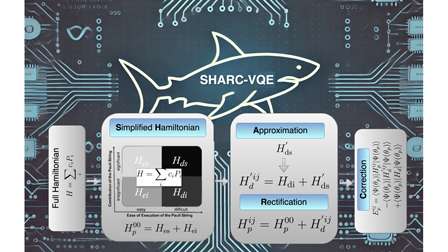
Reducing errors in noisy quantum machines
Perhaps the most striking result is the way SHARC-VQE handles noise. On current NISQ devices, standard VQE simulations suffer errors of 30 to 40 percent. Using SHARC-VQE, the same simulations report errors of only 5 to 10 percent, without adding any error-correction layers. This improvement could be transformative for researchers struggling with fragile quantum hardware.
The reduction is achieved because the partial Hamiltonian approach groups easy-to-measure terms into a single circuit, dramatically reducing the number of noisy operations. Instead of scaling as O(N⁴/ε²), the measurement cost collapses to O(1/ε²), a significant leap in efficiency.
Faster simulations with less computational cost
The numbers speak for themselves. Traditional VQEs require computational resources that scale with O(N⁷/ε²). SHARC-VQE reduces this to O(N³/ε²). For larger molecules, this is the difference between simulations that are practically impossible and those that could run on available quantum hardware.
Benchmarks on molecules such as hydrogen (H₂), lithium hydride (LiH), beryllium hydride (BeH₂), and water (H₂O) show that SHARC-VQE consistently converges to the correct ground-state energy within chemical accuracy. Even excited states, typically harder to capture, can be calculated effectively using a variant of the method combined with variational quantum deflation.
Implications for drug discovery
One of the most anticipated applications of quantum chemistry is drug discovery. Pharmaceutical companies spend billions on drug design, with much of that cost tied to simulating how potential molecules interact with proteins in the body. Faster and more accurate quantum simulations could cut development times and reduce reliance on costly laboratory testing.
SHARC-VQE’s ability to handle noisy environments without elaborate error correction means drug discovery teams could begin experimenting with real quantum devices sooner than expected. Simulating binding affinities, electronic structures, or reaction pathways could become feasible for complex molecules that currently push the limits of supercomputers.
Energy and materials applications
The impact extends beyond medicine. Energy researchers are racing to design better catalysts for green hydrogen production and advanced materials for batteries. These materials are often defined by strongly correlated electron systems that defeat classical approximations.
SHARC-VQE offers a route to explore such systems at scale. By simplifying the Hamiltonian while retaining accuracy, the algorithm could help uncover new materials for renewable energy, carbon capture, and superconductivity.
The barren plateau challenge
Variational quantum algorithms are not without pitfalls. One known problem is the barren plateau phenomenon, where the optimisation landscape becomes flat and algorithms struggle to find minima. Simplified Hamiltonians are often suspected to make this worse.
The multi-disciplinary team from Chemistry, Physics, and Data Science Departments at IIT Kharagpur tested SHARC-VQE against this challenge. Surprisingly, replacing the full Hamiltonian with a partial version did not worsen the barren plateau problem. For molecules such as hydrogen and lithium hydride, the potential energy surfaces generated by SHARC-VQE closely matched those from full Hamiltonians near equilibrium geometries.
Initialising complex problems
Another advantage of SHARC-VQE is its role as an initialisation tool. In simulations of the Fermi-Hubbard model, a fundamental framework for studying correlated electron systems, the partial Hamiltonian generated effective starting points for full VQE calculations. This warm-start capability could shorten simulation times dramatically, helping quantum researchers push into more complex regimes.
In this work, we cut complexity, not corners. With SHARC-VQE, we simplify, refine, and correct, thereby delivering sharper, noise-resistant quantum chemistry.
-Prof. Sabyashachi Mishra
How SHARC-VQE compares to other methods
Several strategies have been proposed to reduce the measurement burden in quantum chemistry. Locality-based approximations, basis rotation grouping, and machine learning approaches all offer partial solutions but often at the cost of extra gates or reduced scalability.
SHARC-VQE stands out because it achieves measurement reduction without requiring additional quantum resources. Its scaling advantage, coupled with resilience to noise, makes it one of the most promising methods yet proposed for quantum chemistry in the NISQ era.
Conclusion
The SHARC-VQE algorithm, developed by researchers at the Indian Institute of Technology, Kharagpur, shows that clever partitioning of molecular Hamiltonians can dramatically reduce both computational costs and error rates. By cutting measurement overhead, improving resilience to noise, and offering warm-start initialisation, this method lays vital groundwork for the practical application of quantum computing in chemistry and beyond.
For scientists and industries waiting on the promise of quantum chemistry, SHARC-VQE delivers a clear message: the future may be closer than it appears.
Indian Institute of Technology Kharagpur (IIT KGP) is the first IIT (established in 1951) of Independent India that created a successful chain of IITs and an IIT eco-system. Currently, the institute is celebrating its 75th year.
Reference
Singh, H., Majumder, S., & Mishra, S. (2025). SHARC-VQE: Simplified Hamiltonian approach with refinement and correction enabled variational quantum eigensolver for molecular simulation. The Journal of Chemical Physics, 162(11). https://doi.org/10.1063/5.0249447





